


📢 转载信息
原文链接:http://bair.berkeley.edu/blog/2025/04/08/plaid/
原文作者:BAIR Blog
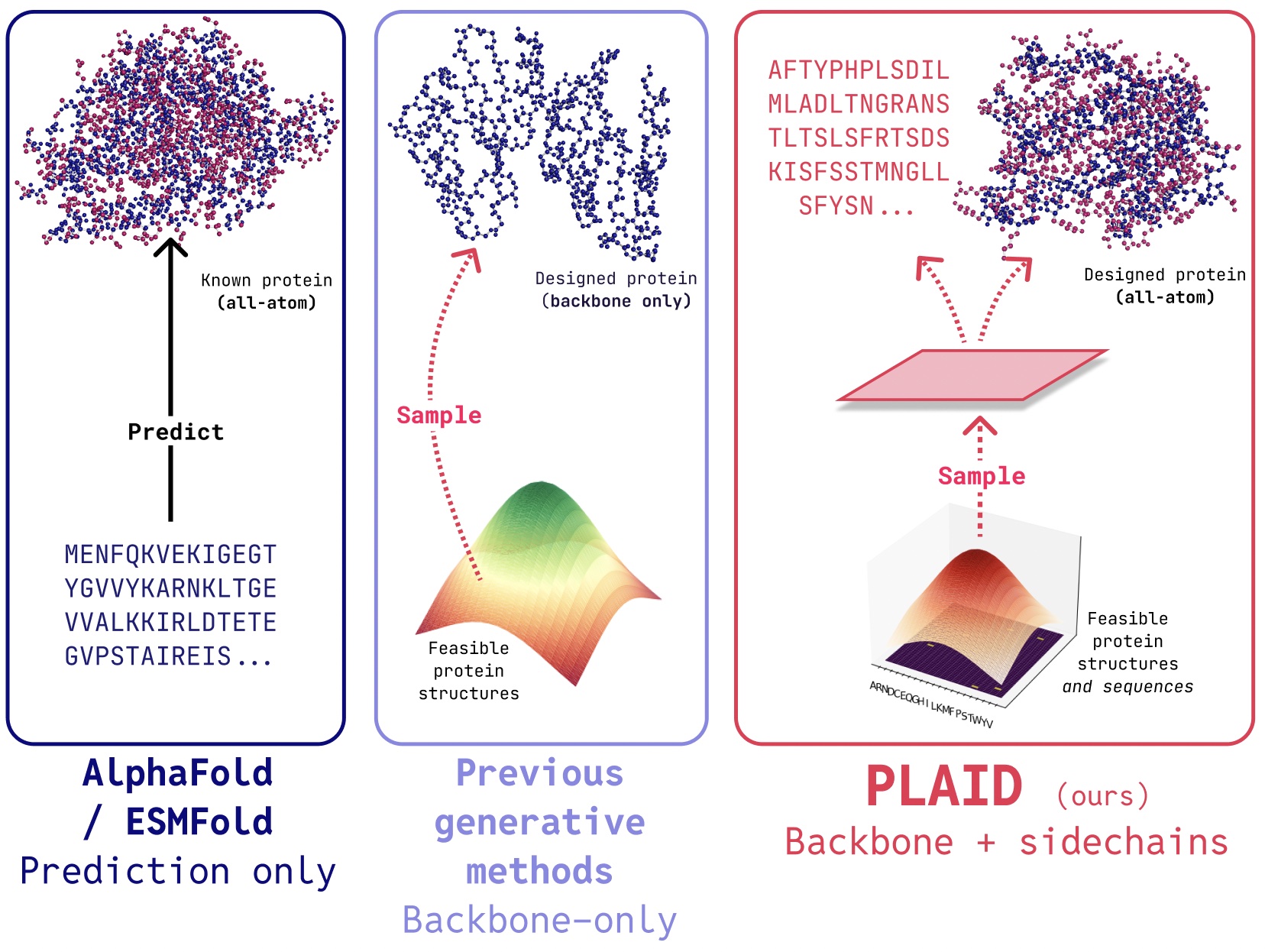
PLAID 是一种多模态生成模型,通过学习蛋白质折叠模型的潜在空间,能够同时生成蛋白质的一级序列和三维结构。
2024 年诺贝尔化学奖授予 AlphaFold2,标志着 AI 在生物学领域的作用得到了重要认可。那么,蛋白质折叠之后还有什么?
在 PLAID 研究中,我们开发了一种新方法,通过采样蛋白质折叠模型的潜在空间来生成全新的蛋白质。它支持复合功能和物种提示词,并能在比结构数据库大 2-4 个数量级的序列数据库上进行训练。与许多以往的蛋白质结构生成模型不同,PLAID 解决了多模态协同生成的问题:即同时生成离散的序列和连续的全原子结构坐标。
从结构预测到现实世界的药物设计
尽管最近的研究表明扩散模型在生成蛋白质方面展现出前景,但现有模型仍存在局限性,使其难以应用于现实场景,例如:
- 全原子生成:许多现有生成模型仅生成主链原子。要生产全原子结构并放置侧链原子,必须已知序列。这产生了一个需要同时生成离散和连续模态的多模态生成难题。
- 物种特异性:供人类使用的生物药物需要进行人源化,以避免被人类免疫系统攻击。
- 控制规范:将药物研发转化为临床应用是一个复杂的过程,如何明确这些复杂的约束条件?例如,在解决生物学问题后,你可能需要考虑药物运输的便捷性,从而增加对溶解度的新约束。
生成“有用”的蛋白质
简单地生成蛋白质不如控制生成过程来获取有用的蛋白质。对此,用户界面应该是什么样的?

受启发于图像生成,我们通过组合文本提示来控制蛋白质生成(示例参考 Liu et al., 2022)。
在 PLAID 中,我们为控制规范设置了类似的接口。最终目标是完全通过文本接口控制生成,在此我们以“功能”和“物种”这两个轴的复合约束作为概念验证:
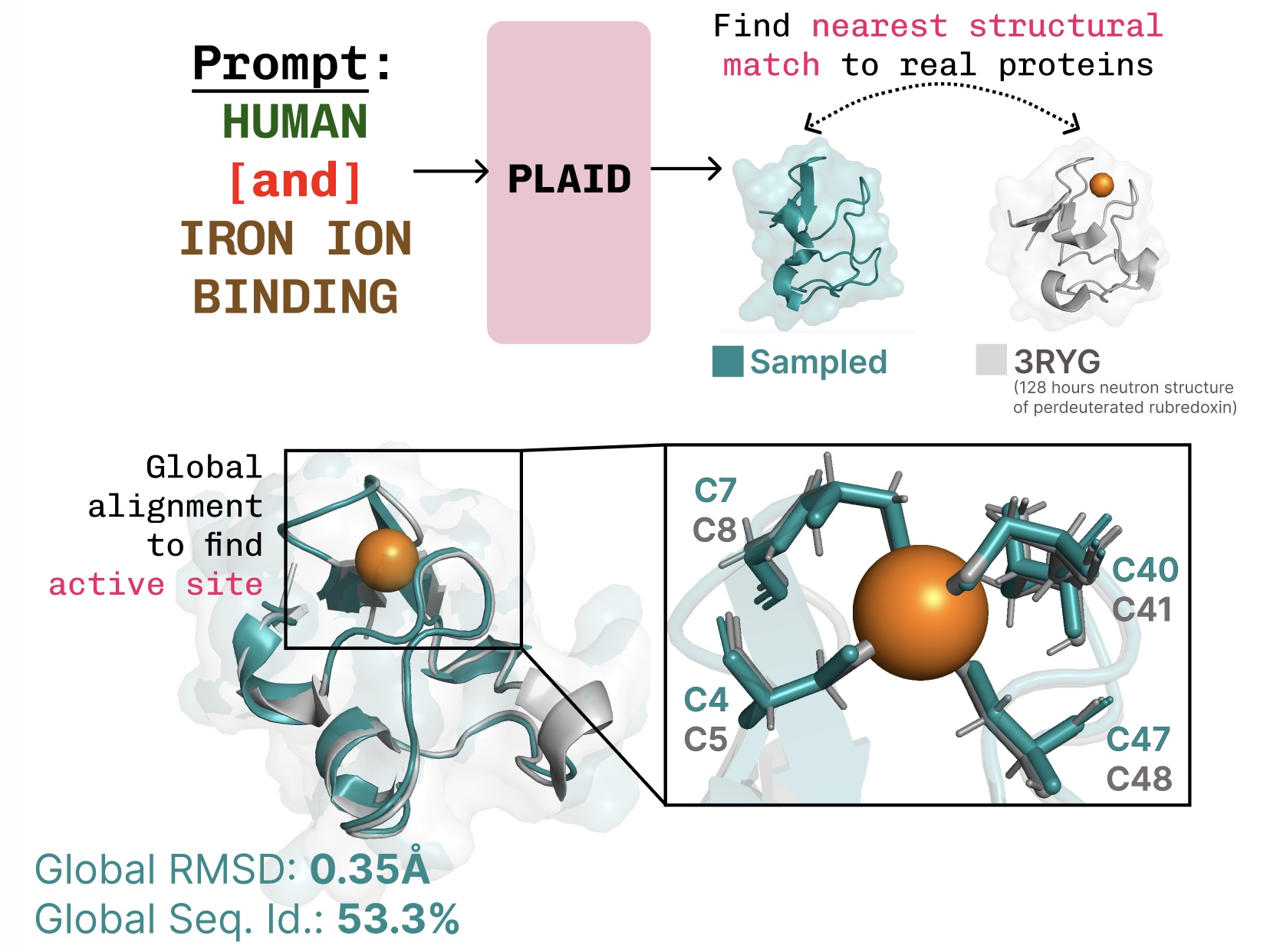
学习功能-结构-序列的联系。 PLAID 学习到了金属蛋白中常见的四面体半胱氨酸-Fe2+/Fe3+ 配位模式,同时保持了高序列水平的多样性。
仅利用序列数据进行训练
PLAID 模型的一个重要方面是,我们仅需要序列数据即可训练该生成模型! 生成模型学习训练数据定义的分布,而序列数据库远大于结构数据库,因为获取序列的成本远低于通过实验确定结构。
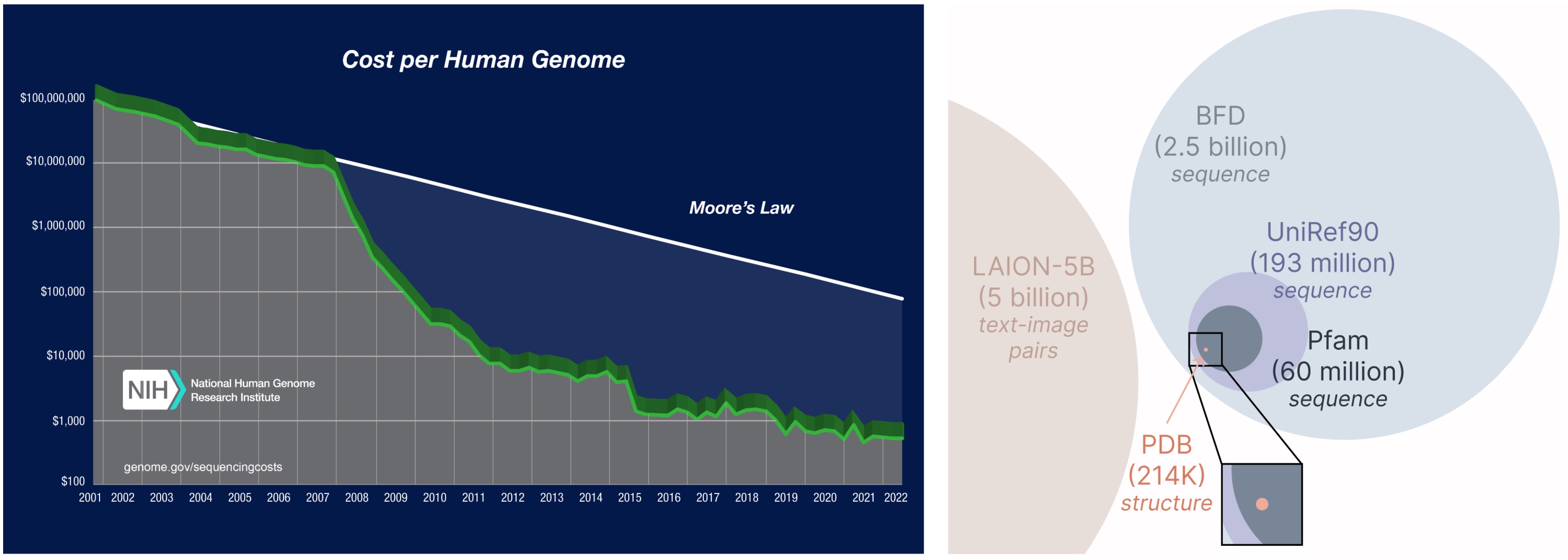
从更大、更广的数据库中学习。 获取蛋白质序列的成本远低于实验测定结构,序列数据库比结构数据库大 2 到 4 个数量级。
它是如何工作的?
我们之所以能够通过仅使用序列数据来训练模型生成结构,是因为我们是在蛋白质折叠模型的潜在空间上学习扩散模型。在推理阶段,从该潜在空间采样出有效的蛋白质后,我们可以利用蛋白质折叠模型的冻结权重来解码结构。这里我们使用了 ESMFold,它是 AlphaFold2 的继任者。
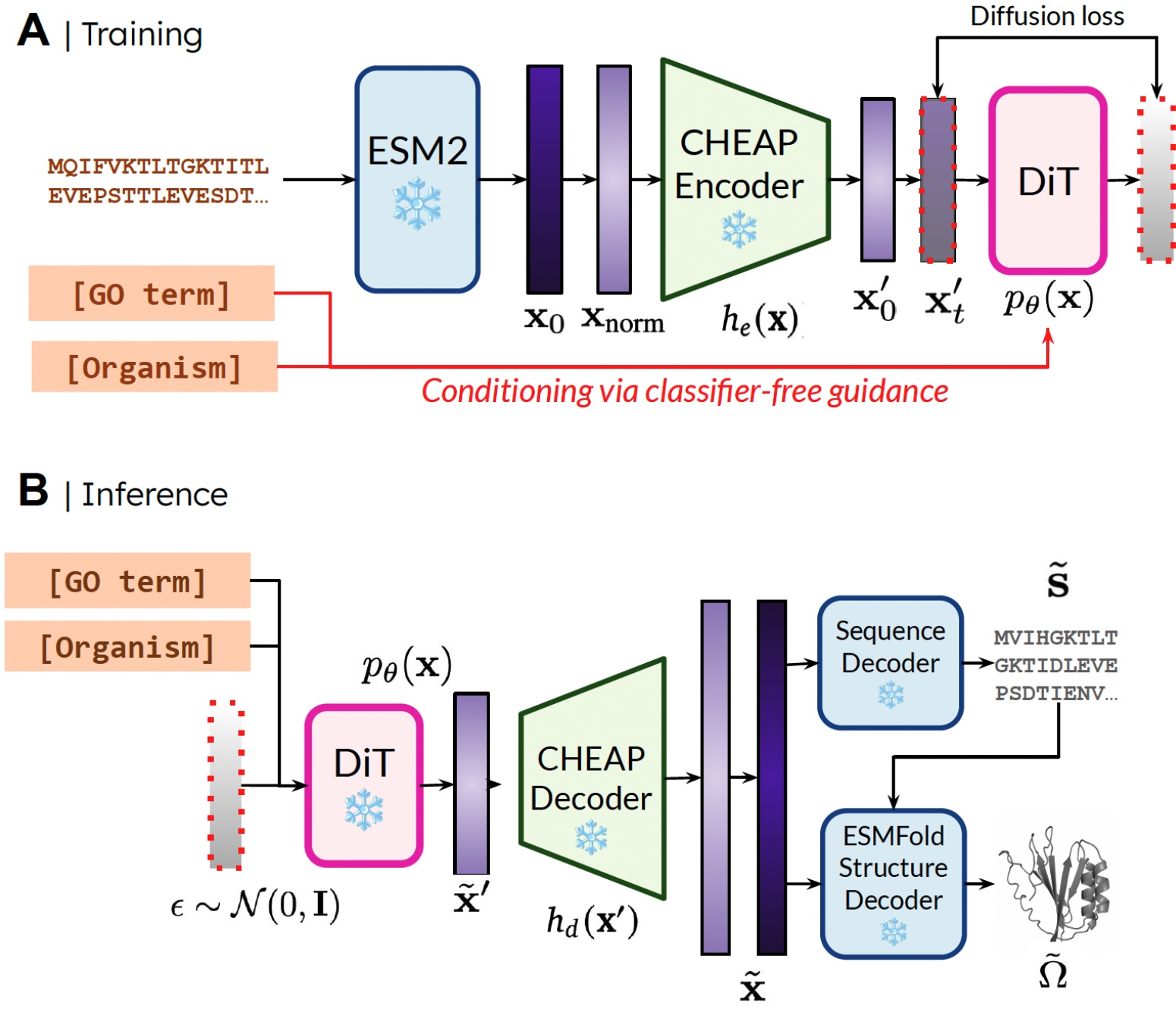
我们的方法。 训练期间,仅需序列即可获得嵌入;推理时,我们可以从采样的嵌入中解码序列和结构。❄️ 表示冻结权重。
压缩蛋白质折叠模型的潜在空间
直接应用该方法的一个小难点是,ESMFold 的潜在空间需要大量正则化。为了解决这个问题,我们提出了 CHEAP (Compressed Hourglass Embedding Adaptations of Proteins),通过学习蛋白质序列和结构的联合嵌入压缩模型,我们发现该潜在空间具有高度可压缩性,从而能够实现全原子蛋白质生成。
后续展望
虽然我们在工作中研究了蛋白质序列和结构的生成,但该方法可以推广到任何存在从高丰度模态到低丰度模态映射的领域。随着序列到结构预测器不断挑战更复杂的系统(如 AlphaFold3 对核酸和分子配体的预测),我们可以想象利用相同的方法对更复杂的系统进行多模态生成。如果您有兴趣合作扩展此方法或进行实验室验证,请与我们联系!
🚀 想要体验更好更全面的AI调用?
欢迎使用青云聚合API,约为官网价格的十分之一,支持300+全球最新模型,以及全球各种生图生视频模型,无需翻墙高速稳定,文档丰富,小白也可以简单操作。
评论区